The AP11 project (Proteomic-genomic Nexus) has reached end of development and we are happy to announce the proteomic-genomic nexus tools are available for the general public.
Introductory Product Information
The Proteomic-Genomic Nexus is a software package that is designed to integrate genomic and transcriptomic data generated from next-generation sequencing with proteomic data generated from protein mass spectrometry.
The primary users of this product will be biologists who would like to integrate their genomics and proteomics data, and be able to visualize them. The tool will be deployed in a number of pilot projects in collaboration with several research groups in Australia and internationally. The research domains span across basic science, primary industry, and medical research.
By using the Proteomic-Genomic Nexus, users will be able to co-visualise genomics, transcriptomic and proteomic data using the Integrative Genomics Viewer. We will be able to validate the existence of genes using peptides identified from mass spectrometry experiments. We will use this to verify alternatively spliced genes by searching for peptides that span across exon-exon junctions.
Instructional Product Information
The tools from developed as a part of the Proteomics-Genomics Nexus project allowed us to visualize all the peptides from the proteome of an organism and efficiently analyze the data.
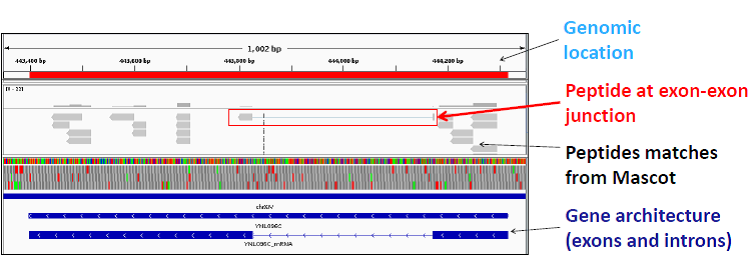
Using the Samifier tool, we co-visualized the genomics, transcriptomics, and proteomics data of Saccharomyces cerevisiae using the Integrative Genomics Viewer (IGV). An example of peptides visualized using IGV is shown in the figure below. The Integrative Genomics Viewer was used to visualize experimental peptides for the yeast 40S ribosomal protein S7-B (YNL096C). A peptide which spans exon-exon junction is highlighted in the red box. This analysis has also been done on a genome / proteome scale.
The Results Analyser was used to verify proteins coded in the Campylobacter concisus (an emergent gut pathogen) and Saccharomyces cerevisiae (Baker’s yeast) genome. Proteins were verified on the basis of two or more peptide ‘hits’, with Mascot scores exceeding an identity threshold. Firstly, for Campylobacter concisus, 66% (1320/2002) of previously known proteins in Uniprot were verified with peptides identified from mass spectrometry experiments. Secondly, for Saccharomyces cerevisiae, 61% (4046/6621) of the proteins as well as 25% (78/284) of all splice junctions in the yeast proteome were verified from a comprehensive proteome analysis (de Godoy et al., 2008, Nature 455:1251-1254). We are in the process of performing analysis on human proteomics data to find evidence for peptides that spans across exon-exon junctions. This will validate the existence of known and novel alternatively spliced transcripts.
Getting started
Please refer to the deployment guide on instructions on how to download or build the tools.
Documentation
There are a number of manual and document resources available:
Github repositories:
Technical documentation:
– AP11 tools - includes links to deployment and developer guides
– Web application - includes links to deployment and developer guides
Overview diagram
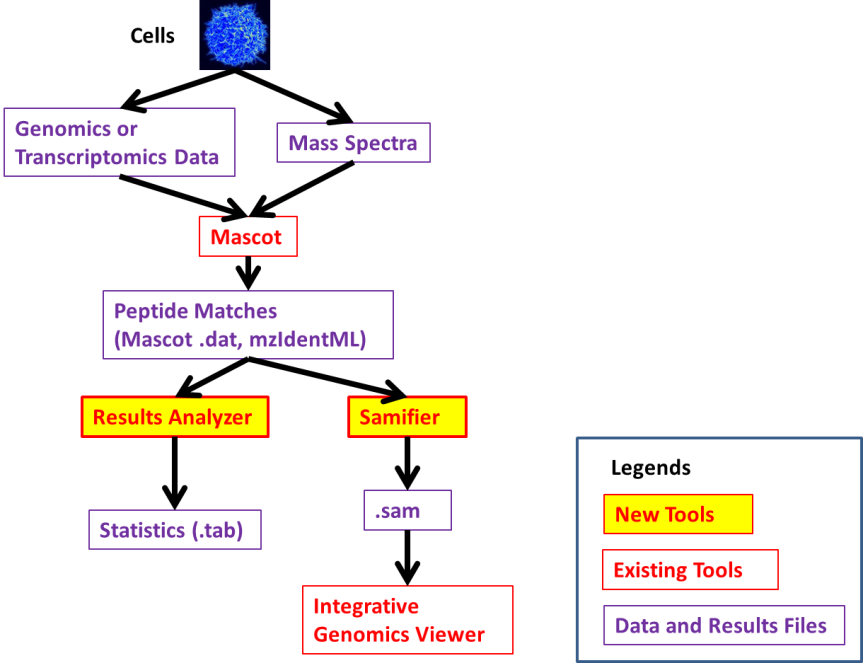
The following diagram (from the wiki,) outlines where the different tools fit within the genomics/proteomics research paths.
Product re-usability information
A number of Java classes where implemented to parse the specific elements of information required by the tools. Their implementation maybe of interest to the genomics and/or proteomics community. They are found in here and include:
A FASTA file reader with caching that can read contig files (see fastaParserImpl.java)
A GFF parser (see GenomeParserImpl.java)
A mzIdentML reader (see mzidentml package), that although specific (it extracts the peptide results required by other parts of the system) is flexible enough to work with version 1.0 and 1.1 of the standard.
There are also a couple of command line utilities
CodonFinder <FASTA-file> <Translation-table> {+|-} [<start>] [<end>]
Prints codons in whole fasta file or between (optional) <start> and <end> parameters. The + or - parameter represents forward or reverse strand translation.
NucleotideToAminoacid <Translation-table> <nucleotide-sequence> [<frame>]
Produces the aminoacid sequence for a given nucleotide sequence, optionally applying a <frame> before translation.
Contextual Product Information
All code is licensed under the GNU GPL v3 license - see LICENSE.txt in each code repository for license text. Documentation (contained in the Github wiki) is licensed under Creative Commons Attribution-Share Alike
Due to the extensive testing by the research stakeholders, the software is now robust, mature and fit for purpose. The ongoing maintenance of the software may occur in a number of ways, as appropriate:
- Intersect provides a 3 month warranty period for the project
- The team at the NSW Systems Biology Initiative may continue to enhance the software
Further enhancements and fixes may be done by Intersect, under the support and maintenance agreement between UNSW and Intersect. T he sustainability of the product has been considered throughout the project and the software has been designed to maximise future maintainability:
The software has an extensive suite of automated unit tests that clearly describe the expected behaviour of the code.
- This is augmented by a suite of integration tests created by the research stakeholders.
- The code is open source so that other groups can contribute code back to the project.
- Using Github makes it easy for changes from other groups to easily be incorporated back into the main code base.
- The system has extensive documentation to explain the design and modules of the system.
- Most of the software was developed in Java as this is the language that the research team at UNSW are familiar with.